The central focus of our research is to understand the pattern and processes of trait evolution through long time scales using phylogenetic trees.
One of the big questions in macroevolution is what drives and maintains biodiversity among clades and through time. This question is frequently addressed by studying the dynamics of diversification; how fast species accumulate and how frequently they go extinct. However, the phenotype diversity of lineages is a fundamental component of biodiversity and cannot be predicted by species dynamics alone. For example, some clades are speciose but lack morphological diversity (e.g., cryptic species) while others show great morphological disparity. Thus, in order to understand macroevolutionary patterns and processes across the tree of life, we need to study the dynamics of trait evolution.
Phylogenies and evolutionary integration among traits
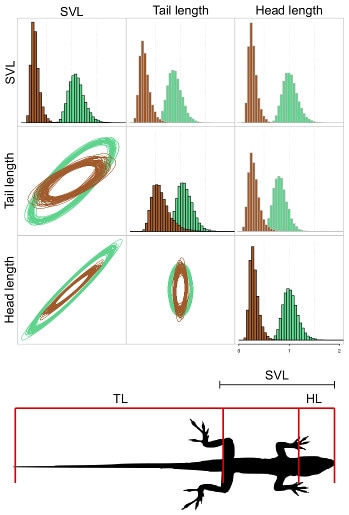
Correlated evolution among traits (known as evolutionary integration) has an important impact on the trajectory of phenotypic evolution. Genetic constraints, ontogeny, and selection have pivotal roles in the development and maintenance of morphological integration over time. Furthermore, shifts in the pattern of evolutionary integration among traits over macroevolutionary scales may play a fundamental role in the evolution of novel phenotypes through exploration of unoccupied regions of morphospace. However, most of what we know about the tempo and mode of trait evolution come from studies that consider only single traits or that use principal component analysis (PCA) to reduce the dimensionality of the data. Studying traits individually excludes the possibility of identifying patterns of evolutionary correlation and the use of PCA does not allow testing for evolutionary shifts in integration. More specifically, we have shown that PCA and phylogenetic PCA (pPCA) can bias the biological interpretation of the mode of evolution underlying the data (Uyeda, Caetano and Pennell, 2015; Systematic Biology) because statistical properties of these methods lead to the first PC axes consistently being estimated as early bursts of differentiation whereas the last axes store a strong signal of stabilizing selection, regardless of the true model.
Therefore, we need models that apply to multivariate data as such in order to better understand macroevolutionary patterns of evolutionary integration. To accomplish that, I have developed a novel framework to facilitate the study of evolutionary integration among traits using phylogenetic trees. The central idea is to estimate rates of evolution for each individual trait in a dataset while also evaluating the structure of evolutionary correlation among them. With this model one can also fit different rate regimes to the phylogenetic tree and detect shifts between regimes due to distinct patterns of evolutionary rates and/or correlations among traits (Caetano and Harmon, under review in Systematic Biology). I implemented this approach in the R package 'ratematrix', that is already available for use (Caetano and Harmon, under review in Methods in Ecology and Evolution – and available at github ).
One of the big questions in macroevolution is what drives and maintains biodiversity among clades and through time. This question is frequently addressed by studying the dynamics of diversification; how fast species accumulate and how frequently they go extinct. However, the phenotype diversity of lineages is a fundamental component of biodiversity and cannot be predicted by species dynamics alone. For example, some clades are speciose but lack morphological diversity (e.g., cryptic species) while others show great morphological disparity. Thus, in order to understand macroevolutionary patterns and processes across the tree of life, we need to study the dynamics of trait evolution.
Phylogenies and evolutionary integration among traits
Correlated evolution among traits (known as evolutionary integration) has an important impact on the trajectory of phenotypic evolution. Genetic constraints, ontogeny, and selection have pivotal roles in the development and maintenance of morphological integration over time. Furthermore, shifts in the pattern of evolutionary integration among traits over macroevolutionary scales may play a fundamental role in the evolution of novel phenotypes through exploration of unoccupied regions of morphospace. However, most of what we know about the tempo and mode of trait evolution come from studies that consider only single traits or that use principal component analysis (PCA) to reduce the dimensionality of the data. Studying traits individually excludes the possibility of identifying patterns of evolutionary correlation and the use of PCA does not allow testing for evolutionary shifts in integration. More specifically, we have shown that PCA and phylogenetic PCA (pPCA) can bias the biological interpretation of the mode of evolution underlying the data (Uyeda, Caetano and Pennell, 2015; Systematic Biology) because statistical properties of these methods lead to the first PC axes consistently being estimated as early bursts of differentiation whereas the last axes store a strong signal of stabilizing selection, regardless of the true model.
Therefore, we need models that apply to multivariate data as such in order to better understand macroevolutionary patterns of evolutionary integration. To accomplish that, I have developed a novel framework to facilitate the study of evolutionary integration among traits using phylogenetic trees. The central idea is to estimate rates of evolution for each individual trait in a dataset while also evaluating the structure of evolutionary correlation among them. With this model one can also fit different rate regimes to the phylogenetic tree and detect shifts between regimes due to distinct patterns of evolutionary rates and/or correlations among traits (Caetano and Harmon, under review in Systematic Biology). I implemented this approach in the R package 'ratematrix', that is already available for use (Caetano and Harmon, under review in Methods in Ecology and Evolution – and available at github ).
|
Evolution of phenotypic sequences
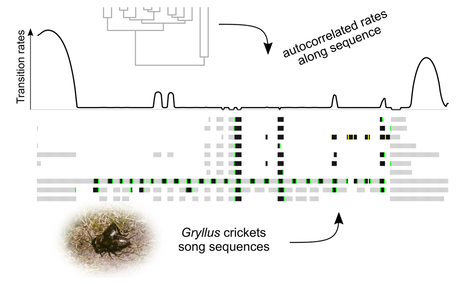
One overarching goal of my research is to expand the type of questions we ask using phylogenies. In the past, most models of trait evolution were developed to study morphological and ecological traits, and few models have been implemented to characterize animal behavior, development, or physiology. It is true that gathering these data can be challenging, but an important bottleneck is that current models have not been designed with these types of data in mind. I described an innovative type of multivariate trait, which I called phenotypic sequence trait (Caetano and Beaulieu, 2019 - American Naturalist - In press). A sequence trait is a series of traits organized in function of some gradient. For example, ontogenetic changes are a series of developmental events organized in time. Similarly, animal behavior is a series of units expressed over time or as a function of the behavioral context. I showed that we can use comparative models to study the evolution of trait organization by quantifying autocorrelation of evolutionary changes (Caetano and Beaulieu, 2019 - American Naturalist - In press). The organization of the traits in a sequence is another dimension of trait integration, because neighboring traits (or traits relatively close to each other) can show strong correlation of rates across the branches of the tree. This new approach to evolutionary integration will significantly impact the kind of macroevolutionary studies we conduct today, by enabling the adequate modeling of a wide range of traits rarely incorporated in comparative studies. |
Models of hidden state geographic evolution (GeoHiSSE)
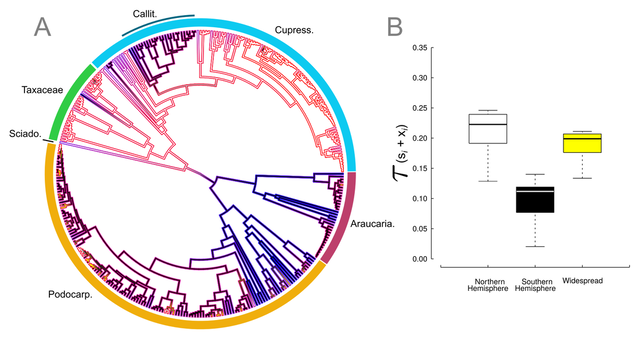
What factors drive the diversification dynamics across clades is a central question in macroevolution. Geographic distribution, together with changes in ecology, species interaction, and dispersion patterns, is one of the most important factors associated with the patterns of speciation and extinction of clades. Goldberg et al. 2011 introduced the GeoSSE model (geographic state-dependent speciation and extinction) to help address these questions. This model incorporates lineage dispersion, speciation by cladogenesis and sympatric speciation as well as area-dependent extinction rates. Unfortunately, the same issue with model inadequacy demonstrated for the BiSSE model (Binary State Speciation and Extinction) is present on the GeoSSE model as originally implemented. Recently, Jeremy Beaulieu, Brian O'Meara, and I published (Caetano et al., 2018 - Evolution) an extensive evaluation of the behavior of the GeoSSE model and introduced the use of hidden states in order to adequatelly take into account diversification shifts that are not associated with patterns of geography. We show that the use of hidden states significantly improve the adequacy of the GeoSSE model. Our implementation, named GeoHiSSE, is available on the 'hisse' package.
What factors drive the diversification dynamics across clades is a central question in macroevolution. Geographic distribution, together with changes in ecology, species interaction, and dispersion patterns, is one of the most important factors associated with the patterns of speciation and extinction of clades. Goldberg et al. 2011 introduced the GeoSSE model (geographic state-dependent speciation and extinction) to help address these questions. This model incorporates lineage dispersion, speciation by cladogenesis and sympatric speciation as well as area-dependent extinction rates. Unfortunately, the same issue with model inadequacy demonstrated for the BiSSE model (Binary State Speciation and Extinction) is present on the GeoSSE model as originally implemented. Recently, Jeremy Beaulieu, Brian O'Meara, and I published (Caetano et al., 2018 - Evolution) an extensive evaluation of the behavior of the GeoSSE model and introduced the use of hidden states in order to adequatelly take into account diversification shifts that are not associated with patterns of geography. We show that the use of hidden states significantly improve the adequacy of the GeoSSE model. Our implementation, named GeoHiSSE, is available on the 'hisse' package.